GSEA Tutorial - GSEA Interface¶
This quick tutorial will guide you through the generation of an Enrichment Map for an analysis performed using GSEA Gene Set Enrichment Analysis directly from the GSEA interface.
Files¶
Download the test data: GSEATutorial.zip
Description of the files contained in the GSEATutorial folder:
ES_NT.clsPhenotype definition for expression file required by GSEA.MCF_ExpMX_v2_names.gctExpression File - Estrogen treatment, Official Gene Name as key. - Data for 12hr,24hr and 48hr.Human_GO_AllPathways_no_GO_iea_April_15_2013_symbol.gmtGene set definition file.
Step 1: Generate GSEA output files¶
- GO to GSEA website
- Click on Downloads in the page header.
- From the javaGSEA Desktop Application right click on Launch with 1 Gb memory.
- Click on “Save Target as…” and save shortcut to your desktop or your folder of choice so you can launch GSEA for your analysis without having to navigate to it through your web browser.
- Double click on GSEA icon you created.
- Click on Load data in left panel.
- Click on Browse for files… in newly opened Load data panel.
- Navigate to directory where you stored tutorial test set files. Select raw expression (.gct) file, sample class file(.cls) and gene set (.gmt) file. Click on Open.
- Wait until confirmation box appears indicating that all files loaded successfully. Click on Ok.
- Click on Run GSEA in left panel.
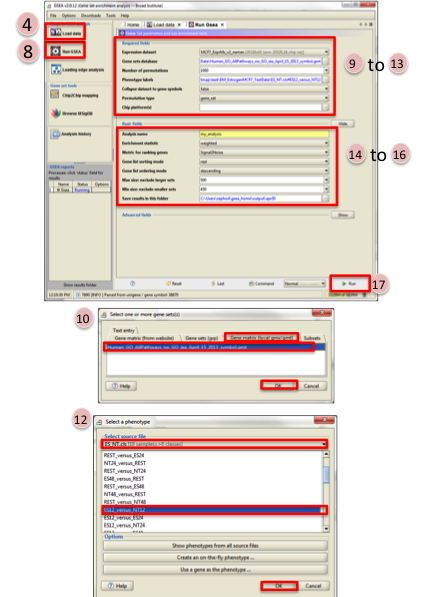
- Select the Expression dataset:
- Click on the arrow next to the Expression dataset text box.
- Select the expression set you wish to run the analysis on (MCF7_ExprMx_v2_names.gct).
- Select the Gene Set Database:
- Click on next to the text box of Gene Set Database.
- Click on Gene Matrix (local gmx/gmt) tab.
- Select gmt file Human_GO_AllPathways_no_GO_iea_April_15_2013_symbo.gmt and click on Ok.
- Select the Phenotype labels file
- Click on … next to the text box of Phenotype labels.
- Make sure Select source file is set to ES_NT.cls.
- Select ES12_versus_NT12 and click on Ok.
- Click on the down arrow next to the text box for Collapse dataset to gene symbols. Select false.
- Click on the down arrow next to the text box for Permutation type. Select gene_set.
- Click on Show next to Basic fields.
- Click in text box next to Analysis name and rename (example:estrogen_treatment_12hr_gsea_enrichment_results).
- Click on … next to Save results in this folder text box. Navigate to the folder where you wish to save the results (preferably the same directory where all the input files have been saved).
- Click on Run in the bottom right corner.
Step 2: Generate Enrichment Map¶
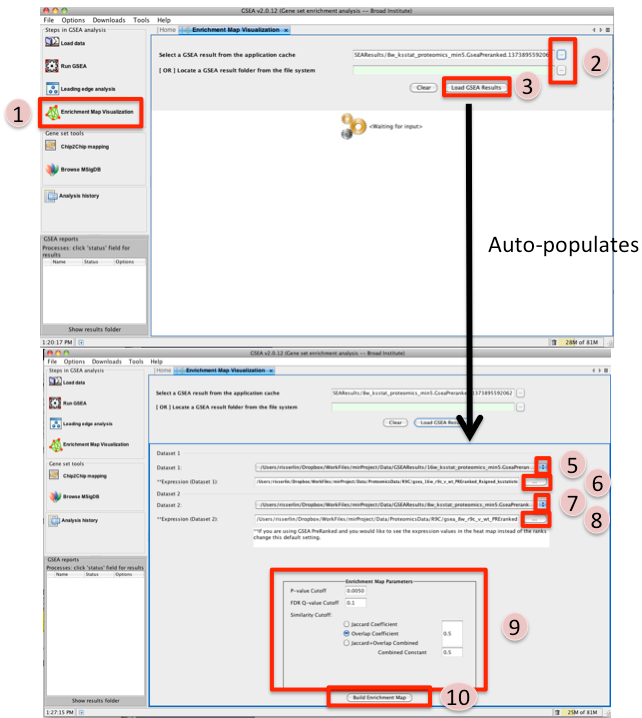
- Once GSEA has completed click in the Steps in GSEA analysis panel click on Enrichment Map Visualization. When you click on the Steps in GSEA analysis cytoscape 3.3 or higher should automatically be launched. It will take a few seconds for cytoscape to load. If you try and create a network before it is finished initializing GSEA will not be able to communicate with cytoscape yet. (In Cytoscape 3.3.0 the Cyotscape splash screen will not disappear until it has finished initializing but in later version you will be able to configure the Enrichment map parameters within GSEA while cytoscape is initializing).
- Navigate to the analysis or analyses you wish to create an enrichment map for. There are
two ways to do this:
- click on the … next to Select a GSEA result from the application cache. From the list select the set of analyses to load. (hold down CTRL or COMMAND key to select multiple analyses). Click on OK. B. click on the … next to [OR] Locate a GSEA result folder from the file system. Navigate to the GSEA result directory you wish to use. Click on OK.
- Click on Load GSEA Results. NOTE: if the GSEA analysis was performed on a dataset that was not collapsed it will take a few seconds for the information to load as it needs to collapse it first.
- Bottom frame will appear, auto-populating file fields according to the GSEA results folders specified. Multiple GSEA folders can be specified. If more than one folder is specified bottom frame will contain specifications for two datasets. If only one directory is specified then only one dataset will be accommodated.
- The user can specify which of the datasets to use as dataset 1 by selecting dataset from drop down list. Selecting a different dataset will automatically populate Expression(Dataset 1) with the corresponding expression file.
- If you have conducted a GSEA analysis on a Preranked list of genes but wish to see the original expression file associated with your enrichment map update the path to the expression file next to Expression (Dataset 1).
- The user can specify which of the datasets to use as dataset 2 by selecting dataset from drop down list. Selecting a different dataset will automatically populate Expression(Dataset 2) with the corresponding expression file.
- If you have conducted a GSEA analysis on a Preranked list of genes but wish to see the original expression file associated with your enrichment map update the path to the expression file next to Expression (Dataset 2).
- Tune Parameters. Check out Tips on Parameter Choice (check out tips for choosing parameters)
- P-value cut-off: 0.001
- Q-value cut-off: 0.05
- Overlap coefficient cut-off: 0.5
- Click on Build Enrichment Map
- Cytoscape should launch and create your Enrichment map.
- Go to View, and activate Show Graphics Details
Step 3: Examining Results¶
Example EM session - Estrogen treatment vs no treatment at 12hr
ES12_EM_example.cys
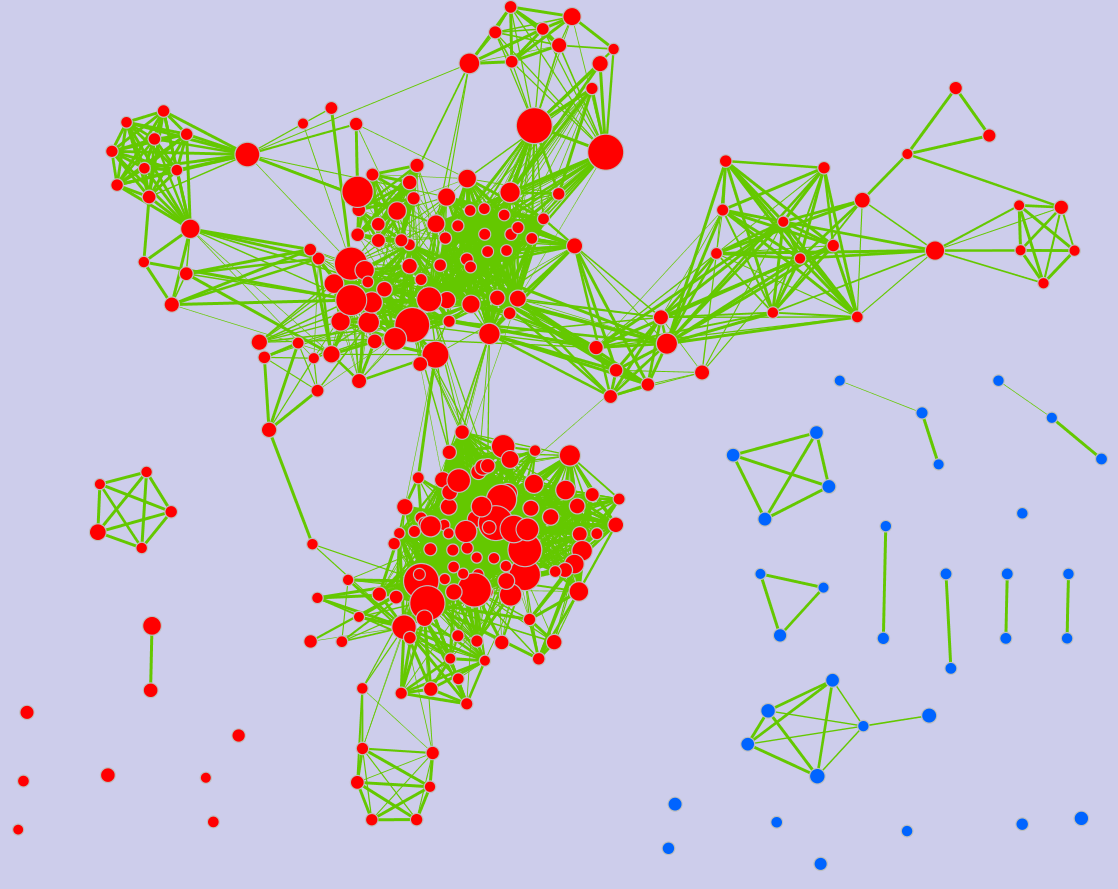
Legend:
- Node (inner circle) size corresponds to the number of genes in dataset 1 within the geneset
- Colour of the node (inner circle) corresponds to the significance of the geneset for dataset 1.
- Edge size corresponds to the number of genes that overlap between the two connected genesets. Green edges correspond to both datasets when it is the only colour edge. When there are two different edge colours, green corresponds to dataset 1 and blue corresponds to dataset 2.
GSEA Leading Edge Information:
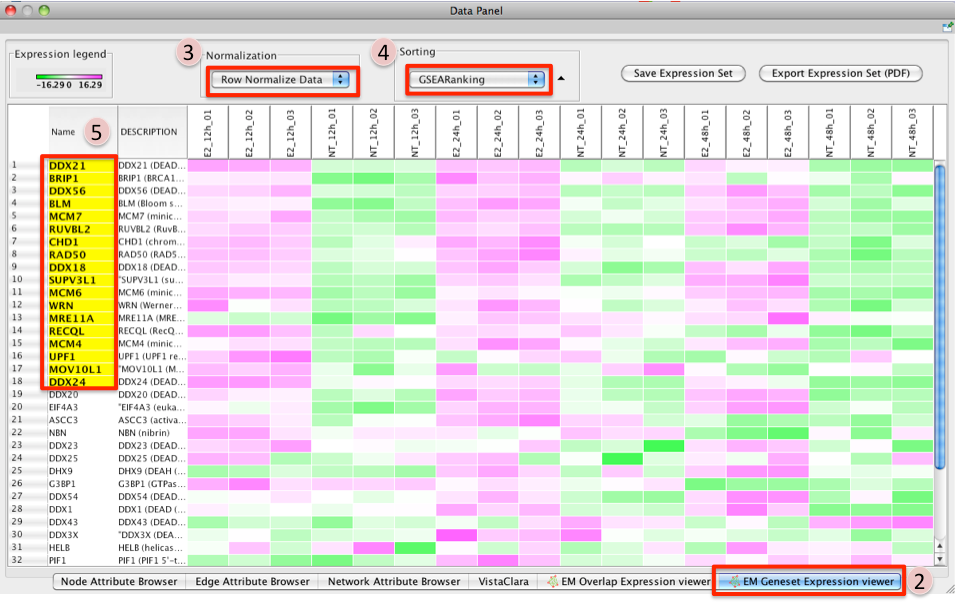
- Click on a node (gene set) in the Enrichment map.
- In the Data Panel, expression profile of all genes included in the selected gene-set should appear in the Heat Map (nodes) viewer tab
- Change the Normalization to your desired metric.
- Change the Sorting method to GSEARanking.
- Genes part of the leading edge are highlighted in yellow.
Note
Leading edge information is currently only available when looking at a single gene set.
More Tutorials¶
For more detailed tutorials check out:
- Merico D, Isserlin R, Bader GD.Methods Mol Biol. 2011;781:257-77. doi: 10.1007/978-1-61779-276-2_12.
- Isserlin R, Merico D, Emili A.Methods Mol Biol. 2013;1005:53-66. doi: 10.1007/978-1-62703-386-2_5.