DAVID Tutorial¶
This quick tutorial will guide you through the generation of an Enrichment Map for an analysis performed using DAVID Functional Annotation Tool,
Files¶
Download the test data: DavidTutorial.zip
Description of the files contained in the DavidTutorial folder:
12hr_topgenes.txtList of top genes expressed in Estrogen dataset at 12hr - Official Gene Symbol.24hr_topgenes.txtList of top genes expressed in Estrogen dataset at 24hr - Official Gene Symbol.12hr_David_Output.txtEstrogen treatment - 12hr DAVID result chart - Screen shot of where to get DAVID output chart24hr_David_Output.txtEstrogen treatment - 24hr DAVID result chartEstrogen_expression_file.txtExpression File - Estrogen treatment, Official Gene Name as key.
Step 1: Generate DAVID output files¶
- GO to DAVID website - http://david.abcc.ncifcrf.gov/
- Select and copy all genes in the tutorial file 12hr_topgenes.txt
- In Upload tab of DAVID interface Paste genes in text box marked Step 1: Enter Gene list
- Select Official Gene Symbol in Step 2: Select Identifier
- Select Gene list in Step 3: Select List Type
- Click Submit list
- Select species: Homo sapiens
- Click Functional Annotation Chart - Screen shot of where to get DAVID output chart
- Download file - This is the file you can use in Enrichment Map (Dataset 1 or 2:Enrichment Results)
Note
Repeat these steps for the 24hrs time-point and the file 24hr_topgenes.txt
Step 2: Generate Enrichment Map with DAVID Output¶
- Open Cytoscape
- Click on Plugins / Enrichment Maps / Load Enrichment Results
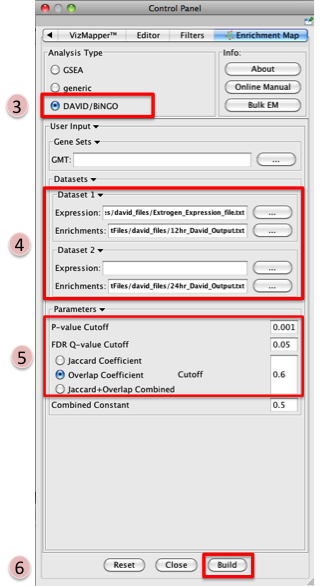
- Make sure the Analysis Type is set to DAVID/BiNGO
- Please select the following files by clicking on the respective (…) button and
selecting the file in the Dialog:
- NO GMT file is required for DAVID Analysis
- Dataset 1 / Expression: Estrogen_expression_file.txt (OPTIONAL)
- Dataset 1 / Enrichments: 12hr_David_Output.txt
- Click on “Dataset 2 arrow_collapsed.gif” to expand the panel
- Dataset 2 / Expression: leave empty
- Dataset 2 / Enrichments 1: 24hr_David_Output.txt (OPTIONAL)
- Tune Parameters
- P-value cut-off 0.001
- Q-value cut-off 0.05
- Check Overlap Coefficient
- Overlap coefficient cut-off 0.6
- Build Enrichment Map
- Go to View, and activate Show Graphics Details
Note
There are multiple values in DAVID that can be used for the Q-value in EM including Bonferroni, Benjamini, and FDR. In EM we use the Benjamini as the Q-value.
Step 3: Examining Results¶

Legend:
- Node (inner circle) size corresponds to the number of genes in dataset 1 within the geneset
- Node border (outer circle) size corresponds to the number of genes in dataset 2 within the geneset
- Colour of the node (inner circle) and border(outer circle) corresponds to the significance of the geneset for dataset 1 and dataset 2, respectively.
- Edge size corresponds to the number of genes that overlap between the two connected genesets. Green edges correspond to both datasets when it is the only colour edge. When there are two different edge colours, green corresponds to dataset 1 and blue corresponds to dataset 2.
Note
If you are using two enrichment sets you will see two different colours of edges in the enrichment map. When the set of genes in the two datasets are different (for example, when you are comparing two different species or when you are comparing results from two different platforms) the overlaps are computed for each dataset separately as there is a different set of genes that the enrichments were calculated on. In this case, since the enrichments were reduced to only a subset of most differentially expressed at each time point the set of genes the enrichments are calculated on are different and overlap are calculated for each set separately.