Heat Map Panel¶
The Heat Map panel shows expression values for genes that are associated with selected nodes and edges.

You may select any number of nodes and/or edges in the network. Selecting an edge is equivalent to selecting the two gene set nodes that are connected to the edge.
Note
“Where are my expression values?”
If there are more than 50 expression values per gene then the Compress: Data Set Median option will be automatically enabled. Set Compress to –None– to see the original values. See below for more details.
Warning
There is a known issue on Mac where sometimes selecting a value in the Compress: combo box has no effect. The workaround is to click on the combo box with the mouse, hold down the mouse button, hover the mouse pointer over the option you want, and then release the button.
Expression Legend¶

Click on an expression value cell in the table to show the Expression Legend. It will not be visible until a value is selected.
Shows the value range and the color gradient for the data set associated with the selected expression value. Note the value range may be different for different data sets.
Toolbar¶

Genes
All
Shows all of the genes from all of the selected gene sets (union).
Common
Shows only genes that are common to all selected gene sets (intersection).
Expressions
Values
Shows the raw values from the expression file(s). Expression values are rounded to two decimal places.
Row Norm
Row normalizes the expression values. For each value in a row of expression the mean of the row is subtracted followed by division by the row’s standard deviation.
Log
Takes the log of each expression value.
Compress
-None-
Shows all of the expression values from the expression file(s).
Class: Median, Min, Max
Only available if a class file was provided when the network was created.
Shows a single column for each class where the value is the median, min or max of the values in the class.
Data Set: Median, Min, Max
Shows a single column for each data set where the value is the median, min or max of all the values.
If the number of expressions per gene is greater than 50 then Compress: Median will be automatically enabled.
Values
When disabled only the color gradients are shown. When enabled the numeric expression values are shown.
Expression values are rounded to two decimal places.
Menu button
Opens the panel options menu.
Table¶

Click on any of the column headers to sort the table by that column.
Gene Column
Description Column
Ranks Column
This column is used to sort by ranks or by hierarchical clustering.
Click the Ranks… button to show a menu of ranking options.

If a data set has a rank file then the ranks will be listed in the menu.
See Panel Menu below for details on how to load additional rank files.
Hierarchical Clustering: Genes are clustered using a hierarchical clustering algorithm based on their expression values, the resulting hierarchy is then used to sort the genes.
Expression Columns
Shows expression values for each experiment.
If there is more than one data set and each data set has common expression values then the values will only be shown once.
If there are two or more data sets and they have different expression values then all the expression values are shown.
Genes that do not have expression data are shown in gray.
A colored bar that runs along the top of the expression column headers can be used to differentiate between the data sets. The color of the bar corresponds to the color shown next to the data set name in the main panel.
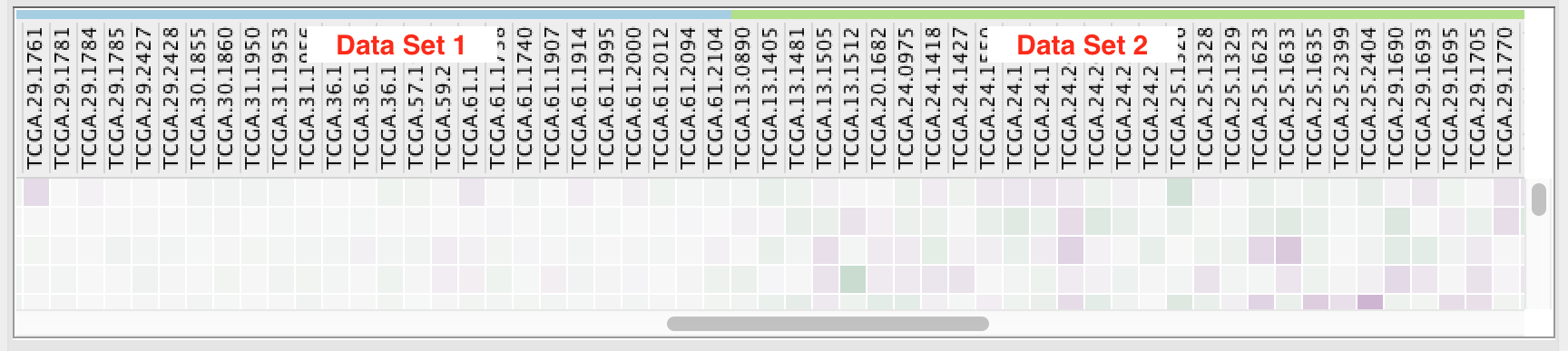
Phenotype Highlight
The phenotypes that were entered in the Create EnrichmentMap Dialog are highlighted.
Leading Edge
Genes that are part of the leading edge are highlighted in yellow.
Available for GSEA results when a single gene set is selected.
See below for more details.
GSEA Leading Edge¶
For every gene set that is tested for significance using GSEA there is a set of proteins in that gene set defined as the Leading Edge. According to GSEA the leading edge is:
“The subset of members that contribute most to the ES. For a positive ES, the leading edge subset is the set of members that appear in the ranked list prior to the peak score. For a negative ES, it is the set of members that appear subsequent to the peak score.”
In essence, the leading edge is the set of genes that contribute most to the enrichment of the gene set.
For Enrichment Map, leading edge information is extracted from the GSEA enrichment results files from the column denoted as Rank at Max. Rank at max is the rank of the gene where the ES score has the maximal value, i.e. the peak ES score. Everything with a better rank than the rank at max is part of the leading edge set.
Panel Menu¶
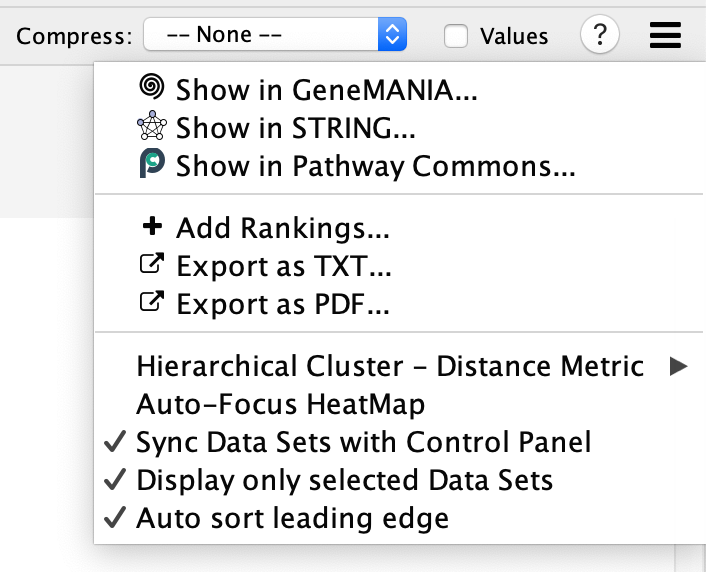
Show in GeneMANIA…
Creates a GeneMANIA network from the list of genes currently selected in the Heat Map.
See GeneMANIA for more details.
Show in STRING…
Creates a STRING network from the list of genes currently selected in the Heat Map.
See STRING for more details.
Show in Pathway Commons…
Opens a web browser and creates a network on the Pathway Commons painter website.
See Pathway Commons Painter for more details.
Add Rankings
Opens a pop-up dialog that allows you to load an additional rank file.
See Add Ranks Dialog below for more details.
Export as TXT
Export the expressions currently being viewed in the heat map table as a tab-separated text file. The first line of the file contains the table headers.
If the heat map is showing the leading edge then you will be prompted to save just the genes that are part of the leading edge or all the genes.
Export as PDF
Export the the expressions currently being viewed in the heat map table as a PDF file.
The visual state of the table is reflected in the PDF file. For example to show the expression values in the PDF file enable the show values option in the toolbar.
Hierarchical Cluster - Distance Metric
Allows to select the distance metric used by the hierarchical cluster algorithm.
Auto-Focus HeatMap
If enabled then every time a node/edge is selected the HeatMap panel will be brought to the front.
Disabled by default.
Sync Data Sets with Control Panel
If enabled then data sets that are not selected in the Control Panel will not be shown in the Heat Map.
Enabled by default.
Display only selected Data Sets
If enabled then data sets that do not contain the gene sets represented by the selected nodes/edges will not be shown in the Heat Map.
Enabled by default.
Auto-sort leading edge
If enabled then the ranking column will automatically sort the leading edge to the top.
Enabled by default.
Add Ranks Dialog¶
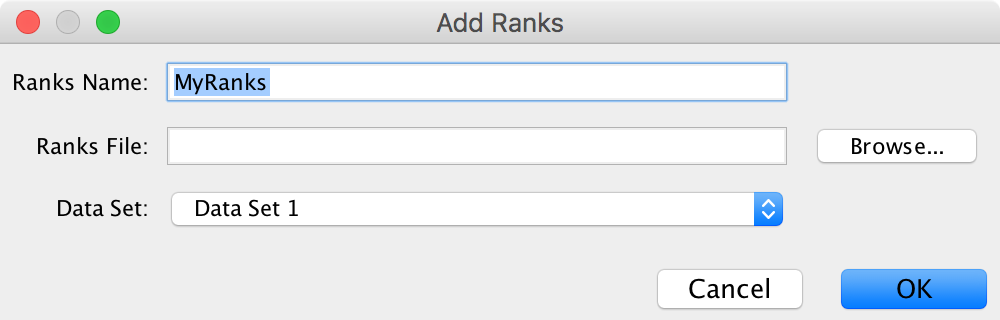
Used to load additional ranks files into an existing data set.